数十年にわたる研究と臨床経験を経て、ムコ多糖症(MPS)の至適管理は新たな時代を迎えています。ムコ多糖症に対する標準治療は現在急速に発展しつつありますが、治療のよりどころとなるのは患者中心の医療(組織化された集学的ケアを実現するヘルスケア提供モデル)において中心的な役割を担う遺伝疾患専門医です。医師にとっては患者さんの生活を変えられる機会です。1-3
ムコ多糖症は多様かつ不定な性質をもつため、組織的なケアを実施するには個々の患者さんに合わせた方法をとり、患者中心の医療を進める必要があります。4 組織的なケアの目的は、患者さんの生活の質を向上させることです。具体的には次のようなものがあります。
ムコ多糖症のような複雑で多臓器にわたる慢性遺伝疾患を持つ小児患者さんの場合、組織化された患者中心の医療によるケアを受けることで、医療機関の利用頻度が少なくてすみ、また健康状態の向上につながります。5-8
患者中心の医療において組織化を行う場合は、より広い範囲のヘルスケアシステム(例.特殊医療、病院、在宅医療、地域サービスなど)を対象とし、患者さんに合わせて作成された管理計画に沿ったものでなければなりません。3
ムコ多糖症の至適管理には、次に挙げる3本の治療の柱があります。適用することによって、患者さんの転帰を向上させることができます。

患者中心の医療を実施するとともに、ERTや長期的なケア、手術ケアに関する管理計画を個々の患者さんに合わせて作成し、最終的には最良の転帰を得られるようにするうえで、呼吸器科医は重要な役割を担うことになります。2,11,13
ERT(可能な場合)は、治療の要となります。12-14
ある患者さんは、進行の速いムコ多糖症VI型を患っていましたが、早い段階で診断が確定されたために、ERTを迅速に開始して臨床転帰を向上させることができました。20

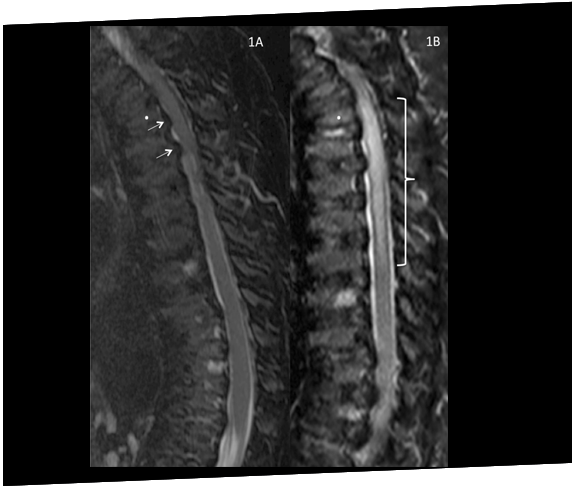
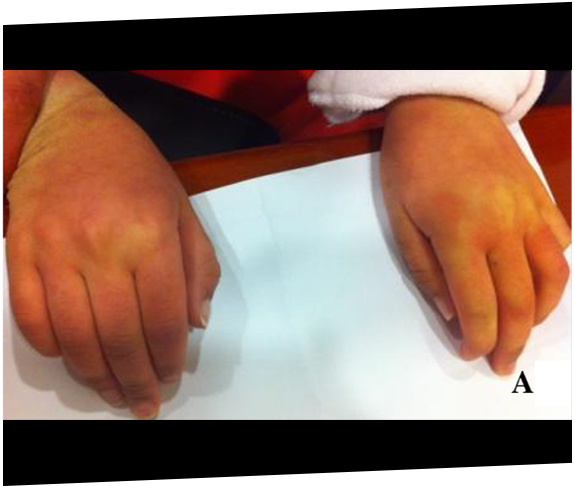
あるムコ多糖症IVA型(モルキオ症候群A型)の患者さんは、外反膝(X脚)の矯正手術を受けた直後に、脊髄圧迫症と対麻痺を経験しました。麻酔中の圧迫に対する脆弱性が分かるケースです。21
ある小児患者さんは、7年間にわたって発育遅延が認められてきましたが、後にムコ多糖症VI型の診断を受けてERTを開始しました。ERTの実施後、臨床的に意味のある改善がみられました。20

ムコ多糖症は臨床的にはっきりと区別できるサブタイプに分けられますが、病態としては共通した特徴があり、どのサブタイプにおいても致死的かつ進行性の多臓器にわたる症状がみられます。14,16,23,24 ムコ多糖症の患者さんを管理するには、具体的な臨床症状とともに、推奨される管理法をサブタイプ別に理解しておく必要があります。2,13
2015年11月
無痛分娩のための両側局所麻酔については、複数回の調整にも拘わらず至適化されず、帝王切開には全身麻酔が必要でした
2015年5月
診断の遅れは、診断施設 がない場合や遠距離にある場合の他、誤診や紛らわしい症状の発現によっても起こりました。症状の発現が予想以上に軽微であったために、見過ごされてしまった患者さんもいました。別の専門領域の観点からムコ多糖症VI型を診断することに関しては特有の課題も浮き彫りになり、 こういった患者さんがどのようにして最初に病院を訪れるのかを知る手がかりを得ることができました。
2016年4月
ムコ多糖症の成人患者さんにおけるその他の心臓関連の問題、特に冠動脈循環と心筋の問題について分かっていることは少なく、この新たに登場してきた成人患者さんたちを効果的にケアするには、さらなる調査・研究が必要とされています。
References: 1. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 3. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 4. Hendriksz CJ, Harmatz P, Beck M, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54-64. doi:10.1016/j.ymgme.2013.04.002. 5. Casey PH, Lyle RE, Bird TM, et al. Effect of hospital-based comprehensive care clinic on health costs for Medicaid-insured medically complex children. Arch Pediatr Adolesc Med. 2011;165(5):392-398. doi:10.1001/archpediatrics.2011.5. 6. Mosquera RA, Avritscher EBC, Samuels CL, et al. Effect of an enhanced medical home on serious illness and cost of care among high-risk children with chronic illness: a randomized clinical trial. JAMA. 2014;312(24):2640-2648. doi:10.1001/jama.2014.16419. 7. Klitzner TS, Rabbitt LA, Chang RKR. Benefits of care coordination for children with complex disease: a pilot medical home project in a resident teaching clinic. J Pediatr. 2010;156(6):1006-1010. doi:10.1016/j.jpeds.2009.12.012. 8. Gordon JB, Colby HH, Bartelt T, Jablonski D, Krauthoefer ML, Havens P. A tertiary care-primary care partnership model for medically complex and fragile children and youth with special health care needs. Arch Pediatr Adolesc Med. 2007;161(10):937-944. 9. Chiang J, Raiman J, Cutz E, Solomon M, Dell S. Tachypnea of infancy as the first sign of Sanfilippo syndrome. Pediatrics. 2014;134(3):e884-e888. doi:10.1542/peds.2013-2765. 10. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12(2):133-138. doi:10.1016/j.prrv.2010.10.005. 11. Berger KI, Fagondes SC, Giugliani R, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):201-210. doi:10.1007/s10545-012-9555-1. 12. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 13. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. 14. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95-101. doi:10.1097/GIM.0b013e3181fea459. 15. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141-1166. 16. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 17. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. doi:10.1007/8904_2014_298. 18. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584. 19. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983. 20. Data on file. Biomarin Pharmaceutical, Inc. 21. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 22. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78. 23. Clarke LA, Winchester B, Giugliani R, Tylki-Szymańska A, Amartino H. Biomarkers for the mucopolysaccharidoses: discovery and clinical utility. Mol Genet Metab. 2012;106(4):396-402. doi:10.1016/j.ymgme.2012.05.003. 24. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 25. Ashworth JL, Biswas S, Wraith E, Lloyd IC. Mucopolysaccharidoses and the eye. Surv Ophthalmol. 2006;51(1):1-17.