
きょうだい症例研究から得られた臨床的証拠によると、早期介入によって疾患特異的な管理とERTの早期開始を行うと、患者さんの転帰を改善する可能性が高くなることが示されています。1-6
ERTは、生後早い時期に開始しても後から開始しても、持久力や肺機能の測定など、生活の質、歩行運動の維持、日常生活動作に重要な意味を持つ主要な臨床パラメータを改善する効果を示しました7,8。
ERTは現在、多くの国でムコ多糖症(MPS)I型、II型、IVA型、VI型の患者さんの治療に用いられています。ムコ多糖症ⅣA型(モルキオ症候群A型)とムコ多糖症VI型に対するERTの有効性のデータと安全性の情報を以下に要約します。
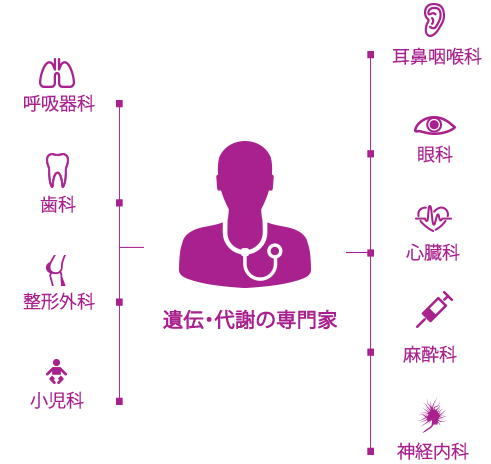
ムコ多糖症(MPS)のような進行性の複合的な遺伝疾患の管理は新たな時代を迎え、患者中心の医療によって各患者さんを担当する医療チームが効率的に連携することが重要です。1
通常は、遺伝疾患や代謝疾患の専門医が患者中心の医療の中心的な役割を果たし、集学的治療と個別化された管理計画の調整をはかることが一般的です。2,3
生涯に渡っての治療方針を考える時、多くのMPSにおいてガイドライン、または専門領域ごとに推奨できる治療法が提供されています。各ガイドラインでは以下のことを推奨しています。2,4
組織化されたケアチームによる早期評価・継続的評価により、患者さんの転帰を向上させることができ、不可逆的な症状の進行の予防にも役立つことがあります。4
例として、下表はムコ多糖症ⅣA型(モルキオ症候群A型)の患者さんに提案される評価スケジュールを表します。2

評価の頻度と特定領域の専門医の関与は、MPSの種類によって異なります。ムコ多糖症I型、II型、III型など原発性の神経変性疾患と認知機能障害を合併するMPS疾患の患者さんは、定期的な神経行動学的および精神医学な評価を追加して実施することが推奨されます。4-6
MPSの患者さんの良好な長期的転帰のために実施すべき専門領域に特化した評価に加えて、全身状態に関連する重要な評価を、担当医師(通常は、遺伝疾患や代謝疾患の専門医)が実施します。疾患および全般的な管理計画について他の医療従事者(歯科医、理学療法士、小児科医、かかりつけ医)や患者ご家族に説明する役割は重要です。これには以下のことが含まれます。2
専門領域に特化した評価、定期的な身体検査、全般的な医療介入は、以下の推奨ガイドラインに沿って実施します。推奨はMPSサブタイプごとに異なる場合があります。2
MPS疾患の治療の向上によって、患者さんの長期的転帰に貢献する上で、生涯にわたる管理への新たなアプローチが必要となっています。
患者さんの年齢が上がるにつれて、一部の患者さんは自分自身で健康管理を始め、医師主導から成人ケアへの移行が不可欠となります。2 医師には以下のことが要求されます。
小児ケアから成人ケアへの移行と長期にわたる成人ケアは、青年期・成人の患者さん向けの治療計画で取り組む重要な領域です。2 長期ケアでの考慮事項は、MPSの症例を多く診療した医療機関で対処するのが理想的であり、複数の専門領域の間で綿密な連携が必要です。2,8 長期的な問題として、例えば以下のことが挙げられます。
継続的な評価と小児ケアから成人ケアへの施設固有の移行戦略などMPS疾患の長期管理は、患者さんの生活の質を長期的に改善し、より良い未来に導く可能性があります。2,8-10
ムコ多糖症(MPS)の臨床症状は多臓器にわたるため、個々の患者さんに合わせて、合併症の発生を予期して管理する集学的アプローチが必要です。1
MPSの患者さんは、生涯に複数回の手術を受けることがめずらしくありません。ムコ多糖症ⅣA型(モルキオ症候群A型)の患者さん325名のコホートを評価する自然経過研究では、患者さんの70%以上が1回以上の外科手術を経験していることが示されました。2

MPSの患者さんは、上気道および下気道閉塞、頸椎不安定症、呼吸障害、心血管疾患、頻回の感染など、複数の要因から周術期の死亡率が高い傾向があります。2-4 例えば、ムコ多糖症ⅣA型(モルキオ症候群A型)の患者さんでは手術合併症による死亡率は11%に上ります(n=27)。5
手術計画の作成は重要であり、(理想的にはMPSの患者さんを治療した経験を持つ)専門医による集学的チームで作成します。3
外科的なリスク評価と周術期モニタリングは、 手術計画の基本要素であり、 これによりMPSの患者さんの望ましくない手術転帰のリスクと死亡率を軽減します。3,9,10


References: 1. McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age—a sibling control study. Clin Genet. 2010;77(5):492-498. doi:10.1111/j.1399-0004.2009.01324.x. 2. Furujo M, Kubo T, Kosuga M, Okuyama T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011;104(4):597-602. doi:10.1016/j.ymgme.2011.08.029. 3. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13-18. 4. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 5. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 6. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95-101. doi:10.1097/GIM.0b013e3181fea459. 7. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 8. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63-72. doi:10.1016/j.ymgme.2013.11.015. 9. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 10. Bagewadi S, Roberts J, Mercer J, Jones S, Stephenson J, Wraith JE. Home treatment with Elaprase® and Naglazyme® is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733-737. doi:10.1007/s10545-008-0980-0. 11. BioMarin Pharmaceutical Inc. VIMIZIM website. http://www.vimizim.com/. Accessed December 21, 2015. 12. BioMarin Pharmaceutical Inc. NAGLAZYME website. http://www.naglazyme.com/. Accessed December 21, 2015. 13. VIMIZIM [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2014. 14. Wood TC, Harvey K, Beck M, et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36(2):293-307. doi:10.1007/s10545-013-9587-1. 15. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 16. Harmatz P, Giugliani R, Schwartz I, et al; for MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or RHASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533-539. doi:10.1016/j.jpeds.2005.12.014. 17. Harmatz P, Giugliani R, Schwartz IVD, et al; for MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94(4):469-475. doi:10.1016/j.ymgme.2008.04.001. 18. Harmatz P, Yu ZF, Giugliani R, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33(1):51-60. doi:10.1007/s10545-009-9007-8.
References: 1. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 4. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. 5. Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Vol 3. 8th ed. New York: McGraw-Hill; 2002:2465-2494. 6. Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi:10.1186/1750-1172-6-72. 7. James A, Hendriksz CJ, Addison O. The oral health needs of children, adolescents and young adults affected by a mucopolysaccharide disorder. JIMD Rep. 2012;2:51-58. doi:10.1007/8904_2011_46. 8. Coutinho MF, Lacerda L, Alves S. Glycosaminoglycan storage disorders: a review. Biochem Res Int. 2012;2012:471325. doi:10.1155/2012/471325. 9. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of Child Neurology. New York, NY: McGraw-Hill; 1996:1141-1166. 10. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48.
References: 1. Wold SM, Derkay CS, Darrow DH, Proud V. Role of the pediatric otolaryngologist in diagnosis and management of children with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2010;74(1):27-31. doi:10.1016/j.ijporl.2009.09.042. 2. Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54-61. doi:10.1016/j.ymgme.2013.01.021. 3. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211-219. doi:10.1007/s10545-012-9563-1. 4. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 5. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. doi:10.1007/8904_2014_298. 6. Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22(9):901-907. doi:10.1111/j.1460-9592.2012.03904.x. 7. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 8. Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi:10.1186/1750-1172-6-72. 9. Solanki GA, Martin KW, Theroux MC, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339-355. doi:10.1007/s10545-013-9586-2. 10. Vitale MG, Skaggs DL, Pace GI, et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. doi:10.1016/j.jspd.2014.05.003. 11. Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012;107:15-24. doi:10.1016/j.ymgme.2012.07.018. 12. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983.


